Texte rédigé en 2016. Pour avoir des informations mises à jour, retrouvez le compte rendu des dernière réunions scientifique dans le menu « La recherche », sous menu « Congrès CDKL5 »
Pour comprendre comment les mutations génétiques peuvent entrainer des conséquences comme celles présentes chez les sujets porteurs de la mutation CDKL5, il est utile de comprendre les bases du fonctionnement des gènes.
Comment fonctionne un gène
L’ADN (acide désoxyribonucléique) constitue la partie principale des chromosomes. Il est présent dans les noyaux de nos cellules, sous forme d’une double chaine. Chaque chaine est formée à partir de molécules d’un sucre (le désoxyribose) lié à 4 maillons différents – les 4 bases : l’adénine, la cytosine, la guanine et la thymine. C’est à partir de la combinaison de ces bases que sont définis nos gènes. Deux chaines sont assemblées grâce à la propriété qu’ont les bases à s’apparier 2 à 2 (adénine avec thymine et guanine avec cytosine).
Un gène est une portion spécifique d’ADN qui contient le code d’une protéine. Il garde en mémoire les informations nécessaires à la synthèse de cette protéine.
Pour que cette protéine soit produite, une empreinte des éléments codants sera réalisée dans le noyau de la cellule, à partir d’ARN (acide ribonucléique). Très proche de l’ADN, il présente certaines différences : c’est une simple chaine, le sucre est le ribose et une de ses bases est différente (la thymine remplacée par l’uracile). Grace à la propriété qu’ont les bases à s’apparier 2 à 2 (adénine avec uracile et guanine avec cytosine), une chaine d’ARN dit messager (ARNm) va être synthétisée et elle sortira du noyau de la cellule vers le cytoplasme.
Là, l’ARNm rejoint une structure appelée ribosome (l’unité de fabrication de protéines), qui va le traduire en protéine. En effet, à une succession de 3 bases (un codon) correspond un acide aminé parmi les 20 que comporte le règne vivant. La lecture de l’ARNm par le ribosome va donc générer une chaine d’acides aminés qui va constituer la protéine. Lorsqu’il rencontrera les séquences UAA, UGA ou UAG, le ribosome arrête la synthèse et libère la protéine. Ces séquences sont appelées codon STOP.
Si le gène est le « plan », alors l’ARNm est le « modèle » qui est tiré du gène et utilisé pour fabriquer la protéine.

Organisation d’un gène
Pour un grand nombre de protéines, le gène est composé d’une succession d’exons et d’introns. Les exons contiennent les éléments codant l’enchainement des acides aminés de la protéine et les introns sont des séquences qui séparent les exons comme les espaces et la ponctuation séparent les mots d’une phrase. Le rôle des introns est encore en grande partie hypothétique. Dans certains cas, ils servent à déterminer quels exons seront utilisés pour faire l’ARNm. Au final, ils sont absents de l’ARNm et ne codent pas la protéine. Mais tous les exons ne codent pas pour la protéine. Au début et en fin de l’ARNm on trouve des exons qui contiennent des éléments qui ne sont pas traduits en protéine (en anglais, UTR= untranslated region). Ces régions servent à contrôler la traduction et à réguler la durée de vie de l’ARNm.
Lors de la fabrication de l’ARNm, les introns sont éliminés et les exons sont assemblés par une opération appelée « épissage » (splicing en anglais). Mais cette opération peut se réaliser de façon différente selon les tissus où l’environnement cellulaire et certains exons ne sont pas inclus dans l’ARNm. On parle d’épissage alternatif. Les divers ARNm obtenus sont qualifiés d’isoformes, (on trouve aussi variants ou transcrits).
Enfin, la régulation de l’expression d’un gène dépend aussi de facteurs épigénétiques. Il s’agit de réactions chimiques qui touchent l’ADN et l’ARN et modifient leurs structures et leurs fonctions. Ainsi, un gène, bien que présent et parfaitement normal, peut ne pas être exprimé selon la cellule, sa localisation, l’environnement, l’état physiologique du sujet, voire son état psychologique.
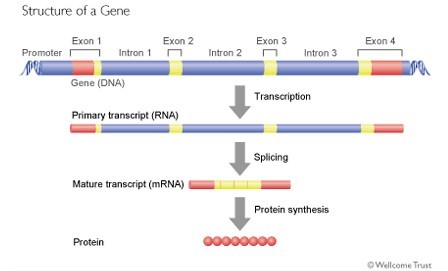
Cas du gène CDKL5
Le gène CDKL5 est connu depuis 1998[1]. Il est situé sur le chromosome X. Il contient 24 exons, mais les 3 premiers exons (1, 1a et 1b) et une partie de l’exon 2 sont des régions non traduites en protéine. A ce jour, on a trouvé 5 isoformes distinctes, exprimées dans une grande variété de tissus (cerveau, testicule, prostate, utérus, poumon). Le cerveau est l’organe où l’expression est la plus forte, et c’est l’isoforme 107 qui est majoritairement exprimée, celle ne possédant pas les exons 19, 20 et 21[2].
Toutes les isoformes contiennent les exons 2 à 11 qui codent pour le domaine kinase de la protéine.

Ref : Kilstrup-Nielsen C. et al. What We Know and Would Like to Know about CDKL5 and Its Involvement in Epileptic Encephalopathy. Neural Plasticity Volume 2012.
L’activation du gène CDKL5 et sa traduction en protéine sont régulés par des facteurs épigénétiques et parmi eux, MeCP2, la protéine mise en cause dans le syndrome de Rett[3].
Fonctions de CDKL5
La protéine CDKL5 (aussi appelée STK9 pour serine/thréonine kinase 9) a donné lieu à assez peu de recherches jusqu’à présent. Comme son nom l’indique, cette protéine fait partie de la famille des Ser/Thr kinases, des enzymes qui branchent un groupement phosphate sur une sérine ou une thréonine (2 acides aminés) de certaines protéines, ce qui a pour effet de modifier la fonction de ces protéines. C’est par ce système que les protéines se déplacent à l’intérieur de la cellule, s’assemblent spécifiquement les unes aux autres, déclenchent ou stoppent leur activité enzymatique.
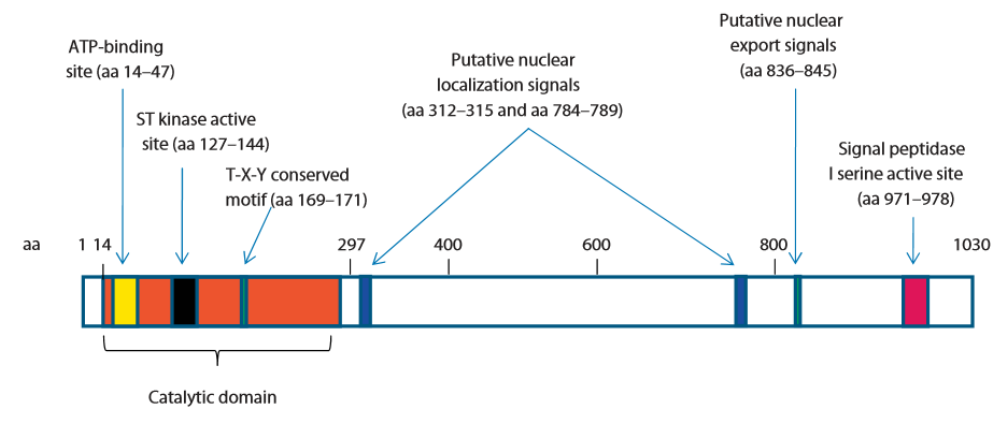
Ref : Bahi-Buisson N et Bienvenu T. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Mol Syndromol. 2012;2(3-5):137-152.
Des études dans des cellules en culture et chez la souris ont permis de confirmer le rôle de CDKL5 dans la croissance et l’organisation des neurones et plusieurs protéines interagissant avec CDKL5 ont été identifiées. Une bonne partie d’entre elles sont connues pour leur rôle dans le développement du système nerveux. Parmi elles, MeCP2, ce qui explique les fortes similitudes entre les sujets CDKL5 et ceux atteints d’un déficit en MeCP2 (Syndrome de Rett).[4], [5], [6], [7], [8], [9],[10], [11], [12], [13], [14],[15], [16].
Mutation de gène
Les mutations sont des modifications irréversibles de l’ADN.
Les mutations chromosomiques correspondent à des cassures et des remaniements de chromosome, ou à des échanges de régions entre chromosomes (translocation). Les conséquences sont très variables suivant l’emplacement et le fait que le gène soit perdu ou non. Les mutations chromosomiques sont détectables par un caryotype.
Les mutations ponctuelles sont des modifications qui touchent un petit nombre de bases, le plus souvent une seule. Elles nécessitent un séquençage pour être identifiées. Elles peuvent être des mutations par substitution (changement de base), des mutations par addition (insertion de base), des mutations par délétion (perte de base).
Une mutation peut ne pas avoir d’effet détectable sur la protéine, soit parce qu’elle se situe dans une zone non traduite, soit que la nouvelle base ne modifie pas l’acide aminé codé (certains acides aminés peuvent être codé par plusieurs codons). On parle de mutations silencieuses. Néanmoins, même si la fonctionnalité de la protéine n’est pas affectée, il est possible que la quantité produite par ces mutants soit moindre.
Lorsque la mutation ne modifie pas le nombre de bases, elle peut soit générer un codon STOP, et raccourcir la protéine (mutation non-sens ou nonsense), soit changer un acide aminé (mutation faux-sens ou misense). Le changement d’un acide aminé peut avoir des conséquences graves, s’il se situe dans une zone importante pour l’activité de la protéine. Mais, dans de nombreux cas, ces variations peuvent être neutre ou de faible effet. C’est ce type de mutation qui génère les différences entre les populations et les individus qu’on appelle polymorphisme génétique. Par contre, la génération d’un codon STOP aura des conséquences plus lourdes, surtout si ce codon survient en début de traduction.
Lorsque la mutation modifie le nombre de base dans la partie codante, le résultat est aussi beaucoup plus radical. En effet, l’ajout ou la perte d’une base va décaler la lecture du code et générer une protéine complètement différente. Dans l’exemple suivant, l’ajout d’une alanine va changer le dernier acide aminé et rallonger la protéine. On obtient des résultats similaires avec une délétion.
5’ AUG CCA UCA GUU UGA CGC UUC CCA UAU AUU AG- 3’
Met-Pro-Ser-Val Stop
5’ AUG CCA UCA*AGU UUG ACG CUU CCC AUA UAU UAG 3’
Met-Pro-Ser-Ser-Leu-Thr-Leu-Pro-Ile-Tyr Stop
Le décalage du cadre de lecture peut aussi générer une séquence de bases codant pour un signal STOP avec pour conséquence un arrêt de la traduction et une protéine tronquée. Qu’elle génère un codon STOP ou des acides aminés inappropriés, si la mutation intervient en début de gène, la conséquence est très importante, car c’est presque toute la protéine qui est affectée.
Si l’addition/délétion porte sur 3 bases consécutives, l’impact peut être moindre car le cadre de lecture n’est pas décalé et ce n’est que l’ajout ou la perte d’un acide aminé.
Mutations sur CDKL5
Entre 2004, l’année de la découverte des premières mutations sur CDKL5 et 2016, environ 200 patients porteurs d’une anomalie ont été clairement identifiés, mais on estime à plus de 800 le nombre de sujets concernés.
Plusieurs facteurs sont à prendre en compte pour les malades
L’emplacement et le type de la mutation
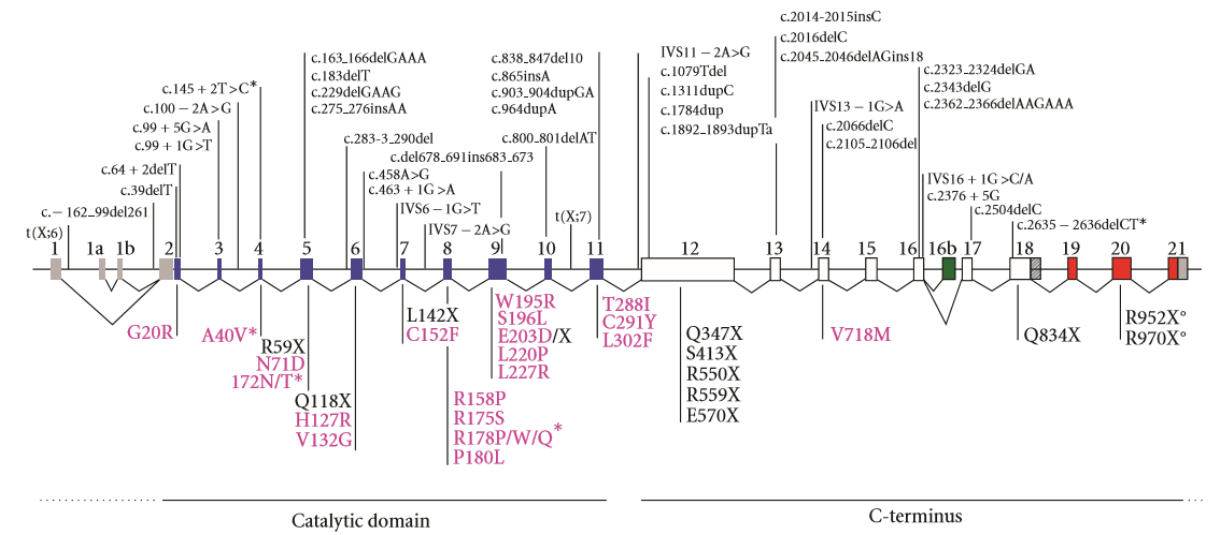
Ref : Kilstrup-Nielsen C. et al. What We Know and Would Like to Know about CDKL5 and Its Involvement in Epileptic Encephalopathy. Neural Plasticity Volume 2012.
Les mutations décrites sont des translocations chromosomiques, des délétions, des insertions, des mutations non-sens et faux-sens. La majorité se situe au début du domaine de la kinase.
Les mutations non-sens et celles décalant le cadre de lecture sont présentes chez les patients les plus sévèrement handicapés[17].
Des tentatives ont été faites pour corréler le type de mutation avec le retard de développement neurologique. Dans une revue très complète parue en 2011, N. Bahi-Buisson et T Bienvenu rassemblent les résultats de 77 cas CDKL5[18]. Parmi ceux-ci, 51 semblent avoir de bonnes compétences motrices, dont 21 cas semblent avoir diverses capacités de marche. L’analyse détaillée des données montre que seulement 30% des personnes ayant une mutation affectant les exons 1 à 11 (ceux qui codent pour le domaine kinase) possèdent une sorte de capacité de marche, alors que ce chiffre augmente à 61% chez ceux qui ont une mutation affectant les exons 12 à 21.
Des résultats allant dans le même sens ont été publiés en 2015, avec 127 cas, montrant que les mutations et délétions qui survenaient après l’acide aminé 781 étaient moins délétères[19]. Néanmoins, des études in vitro ont montré qu’une délétion de la partie située en aval de l’acide aminé 570, même si elle ne provoque pas la perte d’activité kinase de CDKL5 pourrait affecter sa localisation dans la cellule9. D’ailleurs il a été rapporté qu’un patient porteur d’un codon STOP à la place du codon de l’acide glutamique 570 exprime la maladie[20].
Ces études font apparaitre un autre résultat : la maladie chez les garçons est plus rare que chez les filles, mais les atteintes sont plus graves.
Enfin, les mutations sur les 3 derniers exons (19 à 21) ne doivent pas être pathogènes, puisque l’isoforme de CDKL5 exprimée majoritairement dans le cerveau ne les possède pas. Ceci a été vérifié sur 2 sujets[21].
L’inactivation du X
Le gène CDKL5 est situé sur le chromosome X. Ce chromosome est présent en double chez les filles, alors que les garçons en ont un seul, qui est apparié à un chromosome Y, trois fois plus petit. Un seul X est suffisant pour une fonction normale et par conséquent, chez les filles, l’autre est inactivé définitivement durant l’embryogenèse.
A ce moment du développement (environ 1 semaine après la conception), la cellule initiale s’est divisée et l’embryon est formé d’une centaine de cellules. Jusqu’alors, ces cellules sont indifférenciées et elles ont la capacité de donner n’importe quel tissu ou organe. Mais à partir de ce stade, les cellules commencent à se différencier les unes des autres et à se spécialiser pour donner les cellules qui composeront les diverses régions de l’embryon. Jusqu’à cet instant, le choix du X éteint n’était pas définitif, mais il le devient lorsque la cellule se spécialise. Le choix du chromosome X inactivé semble se faire au hasard et il est différent selon les cellules.
En conséquence, à la naissance, chez cette petite fille, les tissus et les organes issus d’une même cellule auront le même X actif et ce chromosome, s’il provient d’une autre cellule, pourra être différent d’un organe à l’autre et même entre des zones différentes d’un même organe.
La mutation CDKL5 n’est généralement présente que sur un des chromosomes X, donc, si dans un tissu ou un organe, c’est le chromosome X avec la mutation qui est éteint, il est possible de n’avoir aucun effet car le chromosome X qui reste actif possède un gène CDKL5 normal.
Il est donc probable que selon la proportion de cellules et leur localisation (cerveau ou autre organe), les effets d’une même mutation soient différents et la pathologie plus ou moins sévère. Ceci laisse entendre que le nombre de sujet porteur de mutation CDKL5 serait beaucoup plus élevé mais que l’expression de la maladie étant moindre ou inexistante, ces sujets ne seraient pas détectés. Chez les garçons, qui n’ont qu’un seul chromosome X, le défaut de CDKL5 est généralement plus dramatique, ce qui peut s’expliquer par le fait que dans toutes les cellules, le chromosome actif est toujours porteur de la mutation.
Comment se produisent les mutations ?
Les mutations peuvent survenir dans n’importe quel type de cellules et sont considérées comme des erreurs de recopiage de l’ADN lors de la réplication des cellules. Leur fréquence est basse (1 à 10 mutants sur 1 million pour les gamètes) et elles sont dues au hasard. A l’origine, l’ADN est abimé lors d’une réplication, de façon spontanée ou sous l’effet d’agents mutagènes (radiations, produits chimiques…). Mais des mécanismes de réparation existent (DNA repair). Ils reconstituent la partie endommagée et restituent un ADN normal. Néanmoins, si les lésions sont nombreuses, elles peuvent ne pas être réparées suffisamment vite et génèrent des mutations.
La présence d’une mutation chez un sujet a deux origines possibles :
Mutation de novo
On parle de mutation de novo lorsque les parents de sont pas porteurs de cette mutation dans leur génome. Cette mutation survient soit au moment de la formation d’un gamète (ovule ou spermatozoïde) chez un parent, soit dans l’œuf nouvellement fécondé, ce qui semble plus rare. La majorité des mutations CDKL5 sont de novo.
Jusqu’à peu, on pensait que le risque de troubles génétiques de novo augmentait avec l’âge de la mère, mais il existe maintenant des preuves que l’âge du père influe tout autant sur la survenue de ces mutations[22].
Mutation héritée
Dans ce cas, c’est un des parents qui est porteur du gène muté dans son génome. En cas de transmission, la mutation sera exprimée si le gène est dominant ou s’il est récessif et que les 2 parents ont apporté le même défaut. Concernant les mutations du chromosome X telles que CDKL5, on peut les considérer comme dominantes, puisqu’un X est inactivé.
Mosaïcisme
Il arrive que des individus n’aient pas tout à fait le même patrimoine génétique dans toutes leurs cellules. Cette disparité est due à des mutations survenant lors de la formation de l’embryon.
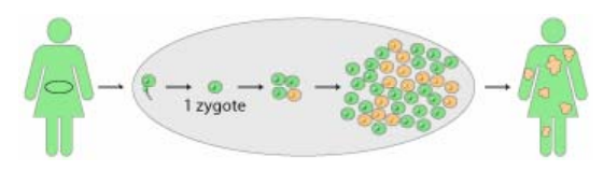
Tout comme l’inactivation du X n’est pas homogène dans toutes les cellules des filles, la présence de cette mutation dépendra du devenir de la cellule mutée dans l’embryon.
MosaÏcisme germinal
Si la mutation touche les cellules germinales donnant naissance aux ovules ou aux spermatozoïdes mais épargne le reste de l’organisme, le parent n’aura aucun trouble même si le gène est fortement affecté. L’enfant, lui, sera porteur.
Dans le cas suivant, une mutation dominante, invisible chez le père, affecte une partie des enfants.
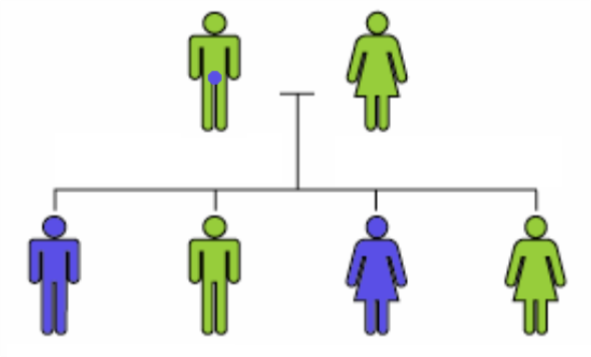
L’analyse sur le génome s’effectuant sur un prélèvement de sang, elle ne pourra pas détecter ce défaut chez le père.
Mosaïcisme somatique
Si une mutation épargne les cellules germinales, la descendance ne sera pas affectée, même si la maladie est exprimée chez le parent.
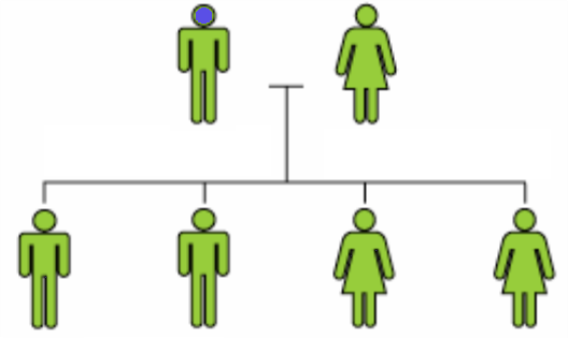
Il est possible que dans certains cas, les mutations CDKL5 puissent être héritées à cause d’une mosaïque des lignées germinales.[23], [24], [25]
Comme on peut le constater, le trouble CDKL5 comprend de multiples paramètres qui peuvent expliquer les différences de symptômes cliniques et leur sévérité et la difficulté qu’il y a à les corréler avec l’emplacement des mutations comme cela a été tenté.
Outre les paramètres génétiques (type de mutation, inactivation du X, mosaïcisme), la prise en charge médicale (traitement, kiné…) doit aussi influencer les symptômes de la maladie.
Devant le faible nombre de cas et compte tenu de la complexité de ces analyses à plusieurs variables, il est important d’alimenter la base de données internationale CDKL5 (CDKL5 International Registry & Database) https://www.cdkl5.com/cdkl5-international-registry-database/. Créée en 2012, elle contient les cas reportés depuis 2007 et elle permet aux chercheurs de confronter leurs hypothèses avec les données cliniques. Elle représente un espoir certain pour la compréhension de cette maladie.
Comment interpréter un rapport d’analyse de gène
Comme nous l’avons vu ci-dessus, une mutation dans une paire de base entraînera une modification de l’acide aminé correspondant et c’est ce changement qui affecte la structure de la protéine CDKL5 et donc sa fonction. Le rapport que vous recevez peut donc faire référence soit à la base qui est affectée (auquel cas la description de la mutation commence par un « c ») soit quel acide aminé a été changé (désigné par un « p »).
Ainsi, par exemple, c.175C>T signifie que la base Cytosine à la position 175 (qui est dans l’exon 5) a été remplacée par Thymine. C’est une substitution.
Un autre exemple serait c.2047delG qui est une suppression de la base Guanine en position 2047 (dans l’exon 14, del comme délétion).
Un autre type de mutation est une insertion. Ainsi, c.865insA signifie que la base Adénine a été insérée dans le gène CDKL5 à la position 865 qui se trouve dans l’exon 11. (Ici, l’exon affecté n’est pas inclus dans le format).
Si le rapport fait référence au changement conséquent dans l’acide aminé, vous verrez quelque chose comme p.Ala40Val, ce qui signifie que l’acide aminé Alanine a été remplacé par l’acide aminé Valine à la position 40 dans la chaîne protéique. Les lettres sont également utilisées pour désigner des acides aminés, de sorte que la même mutation pourrait également être écrite comme p.A40V. Cette modification de l’acide aminé d’Alanine à Valine se produit en raison d’une substitution de base à la position 119 dans l’exon 4 (écrit aussi sous ce format c.119C> T). Dans un rapport, vous pouvez constater que l’un des deux formats est utilisé.
Une protéine tronquée (due soit à une mutation non-sens, soit à une suppression / insertion produisant un codon d’arrêt précoce) est habituellement signifiée avec un « X » comme dans p.R59X qui est dû à la substitution mentionnée ci-dessus, c.175C> T (qui est une mutation non-sens).
Sources
Dr Martyn Newey – http://www.supporting-cdkl5.co.uk/the-genetics-of-cdkl5.php
Bibliographie
[1]Montini E et al. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics. 1998;51(3):427-33.
[2] Williamson SL et al. A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain.Hum Genet. 2012;131(2):187-200.
[3] Carouge D et al. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation.Neurobiol Dis. 2010; 38(3):414-24.
[4] Valli E et al. CDKL5, a novel MYCN-repressed gene, blocks cell cycle and promotes differentiation of neuronal cells. Biochim Biophys Acta. 2012;1819(11-12):1173-85.
[5] Sekiguchi M et al. Identification of amphiphysin 1 as an endogenous substrate for CDKL5, a protein kinase associated with X-linked neurodevelopmental disorder. Arch Biochem Biophys. 2013;535(2):257-67.
[6]Katayama S et al. Critical Determinants of Substrate Recognition by Cyclin-Dependent Kinase-like 5 (CDKL5). Biochemistry. 2015;54(19):2975-87.
[7] Katayama S et al. Expression analyses of splice variants of zebrafish cyclin-dependent kinase-like 5 and its substrate, amphiphysin 1. Gene. 2016;583(1):15-23.
[8] Nawaz MS et al. CDKL5 and Shootin1 Interact and Concur in Regulating Neuronal Polarization. PLoS One. 2016;11(2) :e0148634.
[9] Zhu YC et al. Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc Natl Acad Sci U S A. 2013 ;110(22):9118-23.
[10]Oi A et al. Subcellular distribution of cyclin-dependent kinase-like 5 (CDKL5) is regulated through phosphorylation by dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A). Biochem Biophys Res Commun. 2017;482(2):239-2 .
[11] Fuchs C et al.Inhibition of GSK3ß rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol Dis. 2015 ; 82:298-310.
[12] Amendola E et al. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One. 2014; 9(5):e91613.
[13] Wang IT et al. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc Natl Acad Sci U S A. 2012;109(52):21516-21.
[14] Ricciardi S et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14(9):911-23.
[15] Chen Q et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J Neurosci. 2010; 30(38):12777-86.
[16] Mari F et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005 ; 14(14):1935-46.
[17] Bahi-Buisson N et al. Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am J Med Genet A. 2012;158A(7):1612-9.
[18] Bahi-Buisson N et Bienvenu T. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Mol Syndromol. 2011;2(3-5):137-152.
[19] Fehr S et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology. 2016;87 (21):2206-2213.
[20] Hadzsiev K. et al. Analysis of Hungarian patients with Rett syndrome phenotype for MECP2, CDKL5 and FOXG1 gene mutations. J Hum Genet. 2011;56(3):183-7.
[21] Diebold B. et al.Mutations in the C-terminus of CDKL5: proceed with caution. Eur J Hum Genet. 2014; 22(2):270-2.
[22] Acuna-Hidalgo R. et al. New insights into the generation and role of de novo mutations in health and disease.Genome Biology 2016; 17:241.
[23] Bartnik M et al. Early-onset seizures due to mosaic exonic deletions of CDKL5 in a male and two females. Genet Med. 2011; 13(5):447-52.
[24] Boutry-Kryza N. et al. Complex mosaic CDKL5 deletion with two distinct mutant alleles in a 4-year-old girl. Am J Med Genet A. 2014; 164A(8):2025-8.
[25] Kato T et al. Somatic mosaicism of a CDKL5 mutation identified by next-generation sequencing. Brain Dev. 2015; 37(9):911-5.