Traduction du post de Ana Mingorance, PhD,
http://www.draccon.com/dracaena-report/cdkl5forum2022
Depuis 8 ans, la Loulou Foundation organise une réunion annuelle, le Forum CDKL5, où les scientifiques et les laboratoires pharmaceutique travaillant sur le trouble CDKL5 (CDD – CDKL5 Defiency Disorder), ainsi que des représentants d’organisations de patients, se rencontrent pour discuter des dernières avancées dans le domaine. Vous pouvez trouver les résumés des dernières réunions ici: 2018, 2019, 2020 et 2021.

Le Forum CDKL5 2022 a eu lieu les 7 et 8 novembre à Boston, revenant à un format en face à face après deux éditions en ligne pendant la pandémie. Le fait d’être de retour en personne a rendu la réunion de cette année encore plus spéciale. C’était aussi, à mon avis, la meilleure réunion du Forum CDKL5 auquel j’i pu assister. Notre compréhension de la biologie de la déficience CDKL5 et du rythme de développement du traitement a considérablement progressé au cours des dernières années, je vais donc résumer les principales informations à retenir du Forum de cette année ci-dessous et les comparer à là où nous en étions à Boston en 2019.
1. L’ANNÉE OÙ LE CDD EST DEVENUE UNE PARTIE DES 5 %
On entend toujours la même phrase : « il y a plus de 7 000 maladies rares, et seulement 5 % ont un traitement approuvé ». Il s’agit d’un club spécial de maladies rares qui ont reçu tellement d’attention et d’investissement qu’un médicament a été spécifiquement testé et approuvé pour cette maladie rare.
En 2022, le trouble CDKL5 a rejoint ce club.
En 2022, le Ganaxolone reçoit l’approbation de la FDA (Agence Médicament Américaine) pour le traitement des crises d’épilepsie dans le CDD, et nous attendons bientôt la décision de l’EMA (Agence Médicament Européenne). Lorsque nous nous sommes rencontrés la dernière fois au Forum 2019, nous avons annoncé que Marinus était sur la bonne voie pour terminer le recrutement pour leur essai de phase 3 en cours avec le Ganaxolone pour le traitement de l’épilepsie du CDD. Depuis notre dernière rencontre, Marinus a terminé un tout premier essai clinique mondial pour le CDD au milieu d’une pandémie mondiale, et le Ganaxolone est maintenant disponible pour les patients aux États-Unis et, espérons-le, bientôt dans d’autres pays. Je suis étonné du courage de l’équipe de Marinus de parier sur cette maladie rare et de lutter contre la pandémie pour mettre sur le marché la première thérapie pour les personnes vivant avec le CDD. Ils ont rendu le chemin plus facile à suivre pour tout le monde.
Et pourtant, la première approbation de médicament pour le trouble CDKL5 n’est que l’une des deux plus grandes nouvelles de cette année.
2. LA THÉRAPIE GÉNIQUE CDD ARRIVE AUX ESSAIS CLINIQUES
L’un des plus grands espoirs des familles de patients atteints de CDD est l’arrivée des thérapies géniques dans les essais cliniques. Dans le CDD, la moitié des neurones (chez les filles) ou tous les neurones (chez les garçons) ne produisent pas la protéine CDKL5, qui est une protéine très importante pour le fonctionnement du cerveau. Les thérapies géniques visent à utiliser un virus inoffensif pour transporter une nouvelle copie du gène CDKL5 vers les neurones, afin qu’ils puissent commencer à produire la protéine.

Lors du Forum CDKL5 2022, Emil Kakkis, PDG d’Ultragenyx, a prononcé les mots que les familles CDD attendaient : « La thérapie génique pour CDD arrive à la clinique, nous pensons commencer l’année prochaine ».
Plus tard dans la conférence, Sharyl Fyffe-Maricich (qui a dirigé le programme de thérapie génique CDD chez Ultragenyx) a expliqué toutes les mesures qu’ils ont prises pour optimiser leur thérapie génique afin qu’elle puisse atteindre autant de neurones que possible avant de démarrer les négociations avec l’agence du médicament pour parler du démarrage d’un essai clinique.
Lorsque nous nous sommes rencontrés la dernière fois au Forum CDKL5 2019, nous commencions à disposer de données sur les thérapies génétiques expérimentales chez la souris. Les principales questions à l’époque étaient de savoir si ces thérapies fonctionneraient suffisamment bien chez les animaux pour passer aux essais cliniques, et quand cela pourrait se produire. Nous avons enfin une réponse et un calendrier : si tout se passe bien, les essais cliniques avec la première thérapie génique pour le CDD pourraient commencer dès 2023.
C’était la deuxième grande nouvelle de l’année, et le point culminant du Forum CDKL5 2022.
3.S’ATTAQUER À LA CARENCE EN CDKL5 SOUS TOUS LES ANGLES
Au cours du dîner du Forum, Phil Reilly (un développeur très expérimenté de médicaments pour les maladies rares) a partagé avec nous quelques histoires de maladies qui étaient considérées comme incurables jusqu’à ce que les familles des patients interviennent et jouent un rôle clé dans le développement d’un remède. Son message était le suivant : il n’y a pas de maladie intraitable.
Le lendemain, Emil Kakkis nous rappelait que « le CDD a des composants réversibles, et ce n’est pas vrai pour beaucoup d’autres maladies du système nerveux ». Il faisait référence à ce que nous savons des souris atteintes de CDD, où de multiples aspects de la maladie sont réversibles chez les animaux plus âgés si le protéine CDKL5 est réexprimée. Nous savons également que le CDD n’est pas une maladie dégénérative ou même une maladie neurodéveloppementale où CDKL5 n’est nécessaire que pendant une période limitée de développement. Il s’agit en fait d’un trouble de neuromaintenance, comme nous l’avons vu lors du Forum 2020, il y a donc beaucoup d’espoir clinique autour de la possibilité de restaurer l’expression du gène CDKL5 chez les patients atteints de CDD.
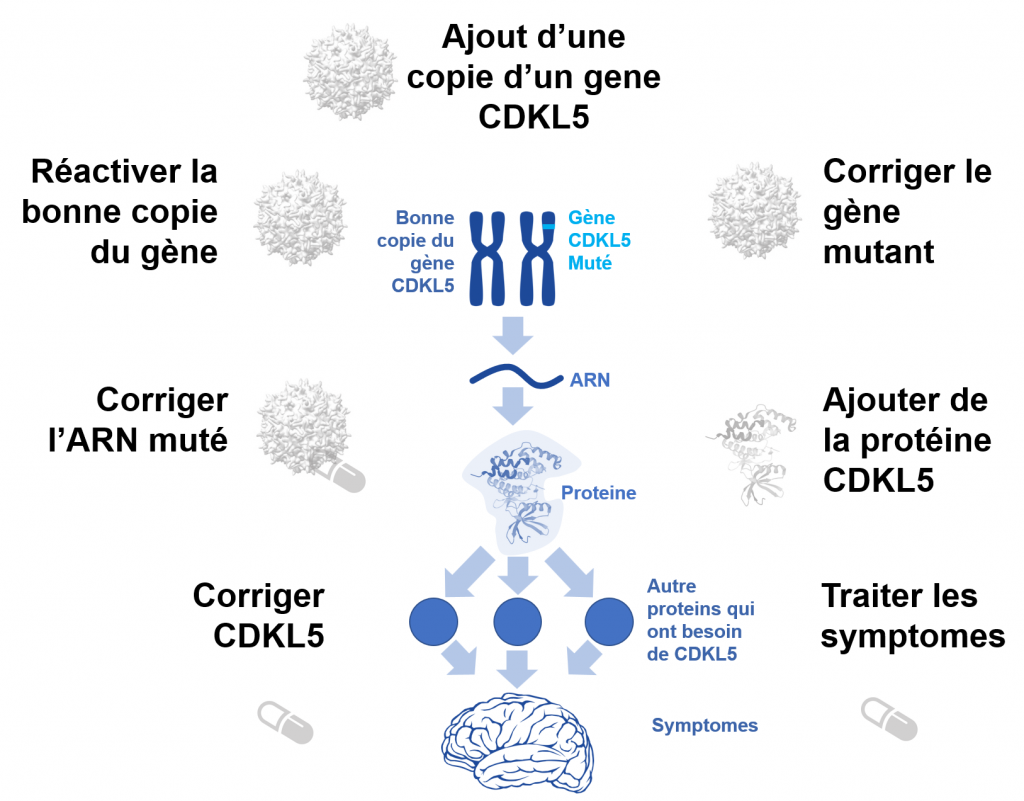
Lors du Forum 2022, nous avons vu de nombreuses mises à jour sur les différentes approches pour lutter contre le trouble CDKL5 sous tous les angles. Voici quelques notes rapides sur les différentes approches:
- Traiter les symptômes de la maladie : un deuxième essai clinique de phase 3 est en cours pour le CDD avec le médicament Fenfluramine. Ce médicament a une bonne efficacité dans le syndrome de Dravet, et la société qui l’a développé (Zogenix) a récemment été acquise par UCB Pharma (développeur du KEPPRA), l’une des principales sociétés d’épilepsie. L’essai clinique est déjà en cours aux États-Unis et s’étendra bientôt à l’Europe et au Japon.
- Traiter les symptômes de la maladie: Le laboratoire Takeda avait évalué l’efficacité et l’innocuité du Soticlestat dans une petite étude sur le CDD et nous avait informés des patients qui avaient continué à prendre le médicament après l’étude. Sur les 12 patients qui ont participé au petit essai, 8 prennent toujours le médicament et tous ont connu une réduction des crises pendant la période où ils ont pris Soticlestat. Et il ne s’agit pas seulement de convulsions, les parents ont signalé des améliorations globales claires, y compris dans la communication. J’espère voir ce médicament envisagé pour traiter les patients CDKL5.
- Corriger le gène CDKL5 : il y a une nouvelle découverte qui ouvre la porte à la conception de nouveaux traitements pour corriger certaines des conséquences directes de l’absence de CDKL5. J’y consacre toute la section 4 de ce compte rendu.
- Ajouter plus de protéine CDKL5: le développement d’une thérapie enzymatique substitutive pour CDD (fabriquer la protéine en laboratoire et la donner aux patients) s’est avéré très difficile. Maria Luisa Tutino a présenté au Forum son travail en vue de produire un gène CDKL5 fonctionnel complet à utiliser pour le remplacement enzymatique. Il est nécessaire que davantage de laboratoires et d’entreprises se joignent aux efforts visant à développer une thérapie enzymatique substitutive pour le CDD parce que c’est nécessaire et très difficile.
- Corriger l’ARNm muté: il existe une variété de stratégies pour essayer de fabriquer une protéine fonctionnelle à partir d’un ARNm muté sans avoir besoin de réparer le gène. Il y a un projet en cours pour développer des oligonucléotides antisens (ASO) qui pourraient corriger les mutations CDKL5 qui causent des problèmes d’épissage (comme Spinraza dans le cas de la maladie SMA) et aussi qui pourraient potentiellement sauter des exons qui pourraient contenir une mutation (sauter une partie de la séquence d’ARN, comme certains traitements pour Duchenne). Il y a aussi des entreprises qui développent des approches pour aider les cellules à lire au-delà d’une mutation non-sens, et j’espère entendre plus de nouvelles sur ces approches et les voir testées pour le CDD.
- Réactiver la bonne copie du gène CDKL5 : nous avons eu deux présentations au Forum 2022 sur les thérapies géniques qui utilisent une version de CRISPR (ciseau génétique) pour trouver la copie CDKL5 inactive et la réactiver. Kyle Fink a dirigé le développement de l’une de ces thérapies géniques pour le CDD au cours des deux dernières années et a récemment reçu une subvention de 1,4 million de dollars pour développer davantage la thérapie génique pour le CDD. Il a également reçu le prix du laboratoire de l’année au Forum CDKL5.
- Corriger le gène muté : il existe plusieurs nouvelles approches pour corriger les lettres dans un gène, ou insérer des lettres. Majid Jafar, co-fondateur de la Loulou Foundation, a expliqué qu’il manquait une lettre à sa fille (Loulou) dans son gène CDKL5 et que sa mère avait demandé « pourquoi ne peuvent ils pas simplement remettre cette lettre? ». David Liu a avancé une approche modifiée de type CRISPR appelée « édition primaire » pour faire précisément cela: remettre une lettre. Il nous a montré comment il a déjà pu réparer le CDKL5 d’enfants comme Loulou dans des cellules en culture, et il travaille maintenant à le faire fonctionner chez des souris vivantes. Certaines de ces approches d’édition de gènes commencent à faire l’objet d’essais cliniques à peine 5 ans après la découverte de la technologie. Son but ultime n’est pas seulement de pouvoir ajouter ou remplacer une lettre, mais de remplacer l’ensemble du gène CDKL5 muté par une copie saine. C’est toujours inspirant et plein d’espoir d’entendre David Liu donner une conférence.
- Ajoutez une autre copie du gène CDKL5: il s’agit de l’approche classique de thérapie génique où les scientifiques utilisent un virus (généralement de type AAV) pour transporter un gène vers les neurones. Le virus ne peut pas se multiplier, il délivre simplement le gène aux neurones et à partir de là, les neurones peuvent produire la protéine manquante, en l’occurrence CDKL5. En 2022, nous avons eu une merveilleuse nouvelle de la part d’Ultragenyx : ils progressent vers des essais cliniques.
4. UNE PERCÉE DANS LA COMPRÉHENSION DE LA PROTEINE CDKL5
Il y a eu de nombreuses présentations au Forum sur la nouvelle compréhension du fonctionnement de la protéine CDKL5 et certaines des conséquences pour le cerveau lorsque CDKL5 est manquant. Mais je n’en soulignerai qu’un dans ce compte rendu, car je considère qu’il s’agit d’une percée majeure dans notre compréhension de la protéine et de la maladie. Il ouvre également une porte importante à de nouveaux traitements.
La découverte est venue du laboratoire de Sila Ultanir, à l’Institut Crick. La protéine CDKL5 est une « kinase », qui sont des protéines qui peuvent modifier la fonction de nombreux autres types de protéines. Cela signifie que lorsque la kinase est manquante, le problème est en cascade, car il y a tout plein d’autres protéines qui ne fonctionnent pas bien. Le laboratoire Ultanir a découvert une protéine très importante qui a besoin de CDKL5 pour fonctionner correctement, et il s’avère être un canal calcique (Cav2.3). Lorsque CDKL5 est manquant, le canal calcique s’ouvre bien, mais se ferme trop lentement, ce qui entraîne une trop grande quantité de calcium.
Ceci est très important parce que toutes les encéphalopathies développementales et épileptiques sont causées par des problèmes avec les canaux ioniques, nous les appelons « channelopathies » en tant que groupe. Par exemple, le syndrome de Dravet est causé par un canal sodique travaillant moins, l’épilepsie KCNQ2 est causée par un canal potassique travaillant trop, et il existe même un syndrome très rare causé par le canal calcique Cav2.3 travaillant trop. La découverte que la carence en protéine CDKL5 fait que ce même canal calcique fonctionne également trop, est inattendue et aide à expliquer beaucoup de choses.
J’appelle cette découverte une percée pour deux raisons principales:
1) On s’est souvent demandé… Si le CDD est causé par une kinase manquante, pourquoi ressemble-t-il autant aux canalopathies? Nous avons maintenant la réponse: CDD pourrait être en partie une canalopathie!
2) Cette découverte ouvre la porte à la conception de nouveaux traitements pour corriger certaines des conséquences directes de l’absence de la protéine CDKL5. Nous voulons maintenant que les entreprises développent des bloqueurs Cav2.3.
5. BEAUCOUP DE PROGRÈS DANS L’ÉLARGISSEMENT DE LA BOÎTE À OUTILS DE RECHERCHE
Pour que les scientifiques comprennent ce que CDKL5 fait dans les cellules et pour tester les traitements des années avant qu’ils ne soient prêts à être mis en essai, ils doivent modéliser la maladie chez les animaux ou les cultures cellulaires en laboratoire. Il y a eu un effort énorme de la part des scientifiques de nombreux pays pour trouver des animaux ou des cellules où nous pourrions supprimer le gène CDKL5 et voir un symptôme fort (que les scientifiques appellent « un phénotype »).
Les animaux les plus utilisés dans la recherche médicale sont les souris et les rats. Cette année, nous avons eu plusieurs excellentes présentations qui élargissent la boîte à outils de recherche aux poissons et aux mouches, et potentiellement aux grenouilles (têtards) et même aux cochons.
Nous avons eu des présentations de deux groupes montrant qu’un petit poisson appelé poisson zèbre pourrait nous aider à rechercher certains des symptômes de CDD. Un groupe du Portugal a constaté que le poisson zèbre manquant de CDKL5 avait des problèmes osseux (ce qui arrive aussi chez les patients!) et des symptômes moteurs intéressants qui pourraient être surveillés pour tester des médicaments expérimentaux chez ces poissons. Et un groupe de Boston est en train de changer la génétique des poissons afin qu’ils puissent manquer de CDKL5 dans seulement la moitié de leurs neurones, ce qui se produit chez les filles atteintes de CDD.

Mes présentations préférées sur les nouveaux modèles CDD :
- Le laboratoire d’Ibo Galindo, en Espagne, a fabriqué des mouches dépourvues du gène CDKL5 et elles ont une épilepsie très forte et meurent même plus tôt que les mouches normales avec CDKL5. En fait, les mouches n’ont qu’un seul gène pour CDKL1/2/3/4 et 5, donc ces mouches manquent de tous les gènes CDKL! Et elles ressemblent à d’autres modèles de mouches ayant une épilepsie génétique. Les mouches ont également des comportements intéressants et pourraient être un système génétique puissant pour comprendre la fonction des 5 protéines CDKL humaines en faisant en sorte que les mouches expriment chacun des gènes humains et voient lesquels peuvent les rendre en bonne santé.
- La société Vyant utilise des cellules souches d’enfants atteints de CDD pour cultiver des organoïdes en laboratoire. Les organoïdes sont des amas de neurones et d’autres cellules cérébrales qui produisent toutes des cellules souches de l’enfant et ressemblent à un mini cerveau dans une boîte de Pétri (ils sont à droite du rat dans les images qui accompagnent ce texte). Il s’avère que les organoïdes d’enfants atteints de CDD ont l’équivalent de l’épilepsie in vitro. Vyant utilise ces organoïdes comme plate-forme pour le ciblage de médicaments directement dans les cellules humaines, et ils trouvent déjà des composés prometteurs. J’ai vraiment aimé leur plate-forme.
J’ai été très impressionné par la ténacité de tant de scientifiques qui font un effort gigantesque pour ne rien laisser au hasard, pour vérifier chaque animal et chaque cellule, les modifier et les optimiser. Cela signifie qu’aujourd’hui, nous avons une variété de modèles différents qui peuvent être utilisés pour répondre à différentes questions sur la biologie et les traitements, et ce type de travail nécessite un gros effort communautaire, il ne peut pas être fait par seulement quelques laboratoires. J’y reviendrai dans la section suivante.
6. « NOUS SOMMES DANS LE MÊME BATEAU »
Un thème qui a émergé lors du Forum CDKL5 2022 était « nous sommes dans le même bateau ». Nous l’avons entendu, et nous l’avons vu en action. Ce ne sont là que quelques-uns des exemples que nous avons vus au cours du Forum :

- Aucun patient n’a été laissé pour compte. Il y a des patients atteints de CDD dans tous les pays. Malheureusement, les barrières linguistiques sont un obstacle majeur pour atteindre la communauté internationale. Afin de rassembler les données du plus grand nombre possible de patients et de pouvoir les contacter au sujet de nouvelles, telles que les essais cliniques. Le registre CDKL5 est récemment disponible dans de nombreuses langues et s’étendra à d’autres. Les langues actuelles sont l’Anglais, le Français, l’Allemand, l’Italien, l’Espagnol, l’Arabe, le Chinois (traditionnel et simplifié), le Japonais et le Russe! C’était beaucoup de travail, mais nous sommes dans le même bateau.
- Entreprises collaborant à des études cliniques. Lorsque nous nous sommes rencontrés pour la dernière fois à Boston en 2019, nous avons parlé de la nécessité d’une étude observationnelle pour valider des mesures précises du CDD (pour vérifier que nous savons comment suivre les symptômes du CDD d’une manière utile pour les essais cliniques). Le hic, c’est que nous ne voulions qu’une seule étude, avec les différentes sociétés pharmaceutiques et biotechnologiques qui se sont réunies pour la concevoir conjointement et la gérer, afin de ne pas surcharger la communauté des patients. Lors du Forum 2022, Xavier Liogier de la Loulou Foundation a annoncé que le premier patient a été recruté dans cette étude, appelée étude CANDID. Les années intermédiaires ont été marquées par beaucoup de travail (y compris l’examen de l’étude par la FDA!) et aussi avec un pas de géant de sept entreprises qui se sont réunies dans une étude qui est une collaboration pré concurrentielle unique en son genre. Nous sommes dans le même bateau.
- Collaborations précliniques. La communauté de recherche du CDKL5 a toujours été exceptionnellement collaborative, et le Forum a une série d’ateliers parallèles (les deux dernières éditions ont également ajouté des ateliers pré-réunion) où nous échangeons. Lors du Forum 2022, nous avons vu une proposition visant à impliquer plus activement les entreprises développant des traitements pour le CDD dans ce réseau de collaboration préclinique, car il est facile pour les scientifiques pharmaceutiques et biotechnologiques de partager des conseils, mais difficile de partager des données de recherche réelles! L’objectif est de permettre aux universitaires et aux entreprises de comparer leurs données de souris CDD pour valider quels sont les critères d’évaluation les meilleurs et les plus fiables (les meilleurs symptômes de souris). Les groupes universitaires le font déjà, et j’espère que d’ici le prochain Forum de l’année prochaine, nous pourrons annoncer que les entreprises se joignent également à cet effort si nécessaire. Nous sommes dans le même bateau.
- L’alliance des patients permet d’accélérer les études cliniques. Un grand défi pour les entreprises de mener des essais sur les maladies rares est de savoir comment trouver les bons hôpitaux. L’Alliance CDKL5 a dressé une liste d’hôpitaux dans les différents pays de l’Alliance où elle a des cliniciens de confiance. Nous avons appris au Forum que cette ressource a déjà contribué à accélérer le lancement de l’étude observationnelle CANDID et j’espère qu’elle contribuera également à accélérer les essais de traitement. Nous sommes dans le même bateau.
- Le leadership des familles de patients. Le Forum CDKL5 est une conférence scientifique et médicale, où nous passons en revue les nouvelles de l’année précédente et nous nous organisons pour aider à faire progresser. Mais c’est aussi une réunion animée par une famille de patients, où les familles des patients figurent en bonne place dans l’ordre du jour et les discussions. Nous avons ouvert la réunion avec les mots de Natalie Ladly de CDKL5 Canada, partageant avec le public le bilan CDD sur sa famille (#brynnstrong!). Simon et Fiona Walsh de CDKL5 Ireland nous ont également invités à examiner la vie de leur famille lors du dîner du Forum, et Rick Upp, de l’IFCR, a clôturé la réunion avec un retour sur la communauté des patients CDKL5. Le prix Champion of Progress CDKL5 de cette année a été décerné à Antonino Caridi, de CDKL5 Italie, l’un des leaders les plus aimés et respectés de la communauté des patients dans la lutte pour un remède contre le CDD. Et des représentants de 14 pays différents (l’Alliance comprend plus de 30 pays !) étaient également présents, se mêlant aux chercheurs et participant activement aux discussions, en particulier lors des séances interactives en petits groupes. La façon dont les mondes et les actions des familles des patients au cours du Forum ont également expliqué la phrase « nous sommes dans le même bateau »

RÉSUMÉ
Si nous devions résumer le Forum à quelques messages, cela ressemblerait à:
- Il n’y a pas de maladie intraitable.
- Le CDD fait maintenant partie des 5% de maladies rares pour lesquels un traitement a été approuvé.
- La thérapie génique CDD arrive à l’essai clinique.
- Et le développement clinique est difficile, mais nous sommes dans le même bateau.
La Loulou Foundation a débuté en 2015 avec la mission d’avoir « des traitements pour le CDD (en essais cliniques) d’ici 2020, et des cures d’ici 2025 ». À l’époque, il n’y avait pas du tout de programmes thérapeutiques en cours d’élaboration pour le CDD, mais il y avait une foi dans la science et dans le pouvoir d’une grande communauté. Le Forum CDKL5 lui-même fait partie de ces efforts, et l’accent mis sur la science et la communauté a porté ses fruits. Avec les nouvelles partagées lors du Forum 2022, il semble maintenant réaliste de croire qu’avant même 2025, il y aura des personnes vivant avec le CDD qui auront reçu une thérapie génique dans le cadre d’un essai clinique. Ce fut une excellente année pour la recherche CDKL5.
J’espère que ce résumé vous a plu. S’il vous plaît laissez-moi savoir vos pensées dans les commentaires.
Ana Mingorance, Ph.D.
Avis de non-responsabilité : ce sont mes propres impressions sur les présentations et les sujets qui m’intéressaient le plus en tant que scientifique et défenseur des patients, et non un texte officiel sur le Forum de la Loulou Foundation. J’écris ces textes en pensant aux parents d’individus avec CDD, donc excusez aussi mon manque de précision technique dans certaines parties.